Innovative Drugs R&D
Our group aims to develop an interdisciplinary platform for innovative drugs research and design. The platform combines the world-leading

Computational Biology
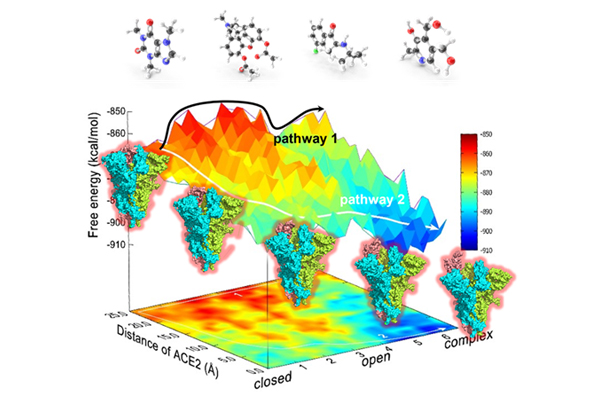
The dynamic characteristics including conformational and energetic information about biological macromolecular are deficient. Professor Arieh Warshel has established a theoretical system of computational biology and been awarded the 2013 Nobel Prize for Chemistry.
One of our goals is to understand the energy mechanisms of various macromolecular machines, especially drug targets, under the guidance of Warshel's theory. And then the molecular mechanisms and dynamic information would serve for drug design.
Currently, we focus on G protein-coupled receptor, SARS-CoV-2 spike, ATPases, ATP citrate lyase, etc.
Read more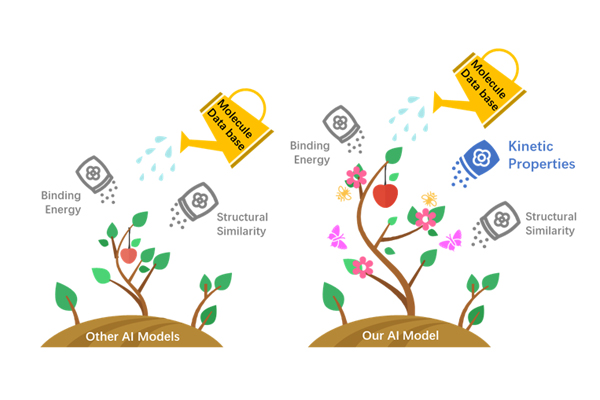
AI Drug Design
Artificial intelligence (AI) has profoundly changed the mode of drug R&D. Compared with traditional methods, AI-aided drug design (AIDD) can explore much larger chemical space, thus improving the structural novelty of drug molecules.
Based on advanced AI drug design algorithms, such as GAN, VAE, and RNN etc., we creatively added kinetic properties of protein functional reactions to AI training.
The kinetic properties more accurately reflect the reality of a protein's functioning. Therefore, we can get the AI drug design model which has the best performance.
Read more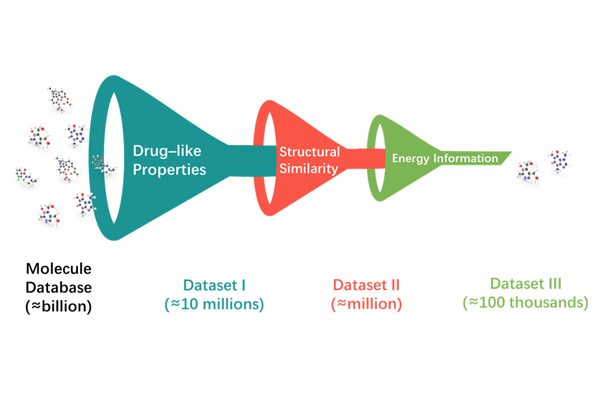
Molecular Virtual Screening
Traversing all molecules in database, whose amount is usually greater than a billion, for drug discovery is a huge temporal and resourceful challenge. Rapid, though crude, virtual screening becomes a viable strategy for reducing the size of molecule library under test.
We use a cascade method to screen the initial molecule database at multiple levels from multiple perspectives, such as drug-like properties, structural similarity, and energy information.
Specifically for energy information, we use a variety of tools for virtual docking of mounts of molecules and target protein pockets to obtain the binding free energy. The docking is performed simultaneously for several key conformations, so we can also obtain the kinetic information.
Read moreCurrent Targets We Studied
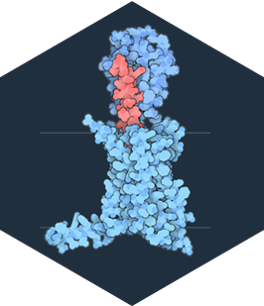
Glucagon Receptor
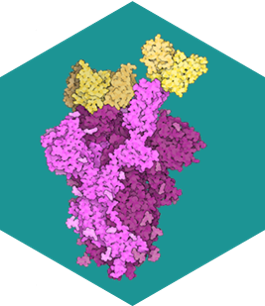
SARS-CoV-2 Spike Protein

ATP-citrate Lyase Enzyme
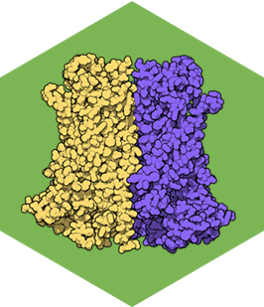
Calcium-activated Chloride Channels
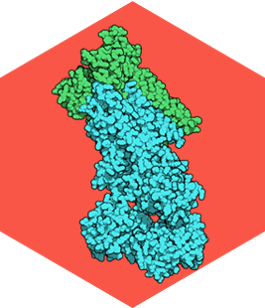
P4 Adenosine Triphosphatases

Soluble Guanylate Cyclase